How should computers think about biology?
Biology is one of the first ways that superintelligence will save (or doom) us all, according to many AI companies. AlphaFold2 won Google the Nobel Prize in 2024 for generating protein structure, which was a longstanding problem in biology. We care about these problems because proteins seem like one of our best paths to cure diseases. If we (or our machine learning models) can intelligently design proteins, it's a step forward towards challenging previously untreatable diseases.
But first, what are proteins? They're the class of molecules behind almost every process in our bodies. Proteins form the capsids protecting viruses on their journeys to infect us, and proteins are the primary operators in our body's immune response. For this reason, they're an important machine learning target. Proteins are composed of a set of building blocks (amino acids) linked together end-to-end like a chain. Surprisingly, just 20 possible building blocks account for the full diversity of tasks that the human body must perform.
If we want to develop models capable of generating new proteins, we'll need to provide them with a language they'll understand. Language models think in tokens, and we typically encode data using a tokenizer. For natural language, this is easy: we use byte pair encoding or something similar to define tokens using subwords. For images, audio, and really everything else, a separate model called a tokenizer compresses continuous data into discrete data which a language model can learn to understand. For this reason, a variety of recent works have proposed tokenizing protein structure and training large, multimodal protein models using structure tokens. These models have performed...okay. Definitely not bad, and they've done some interesting things, but they underperform approaches that skip the tokenization step. The way these models work usually involves pooling information locally. We take a small part of the structure, pass it to the encoder, and extract a vector. Do that for every part of the structure, and now you have an encoding for the whole protein.
This work takes a different approach, where we encode a protein in a way where every token provides global information. The best intuition pump for me is that proteins exhibit all kinds of global properties. Sometimes you interact with one corner of a protein and it has effects on the opposite side of the protein (allostery). Sometimes a small change in one part of a protein completely destabilizes the whole thing. The point is making encodings that are aware of the full protein, rather than just one small snippet, is a very reasonable approach.
In addition to being global, our tokens are adaptive. You don't need every token to encode the protein, you can keep more tokens for more complicated proteins and fewer tokens for simpler proteins. During training, we stochastically mask out tokens and ask the model to regenerate the full structure. The model always has the first token, almost never has the last token, and has the tokens in the middle to varying degrees. It learns to stuff the most important information into the first token (the model always has the first token) and places higher and higher frequency information into later tokens (which the model has less often). I'm using the word frequency deliberately here, and you can make this concrete by thinking of the frequency as the spatial variation over the sequence axis. Take a look at the reconstructions below to see what I mean.
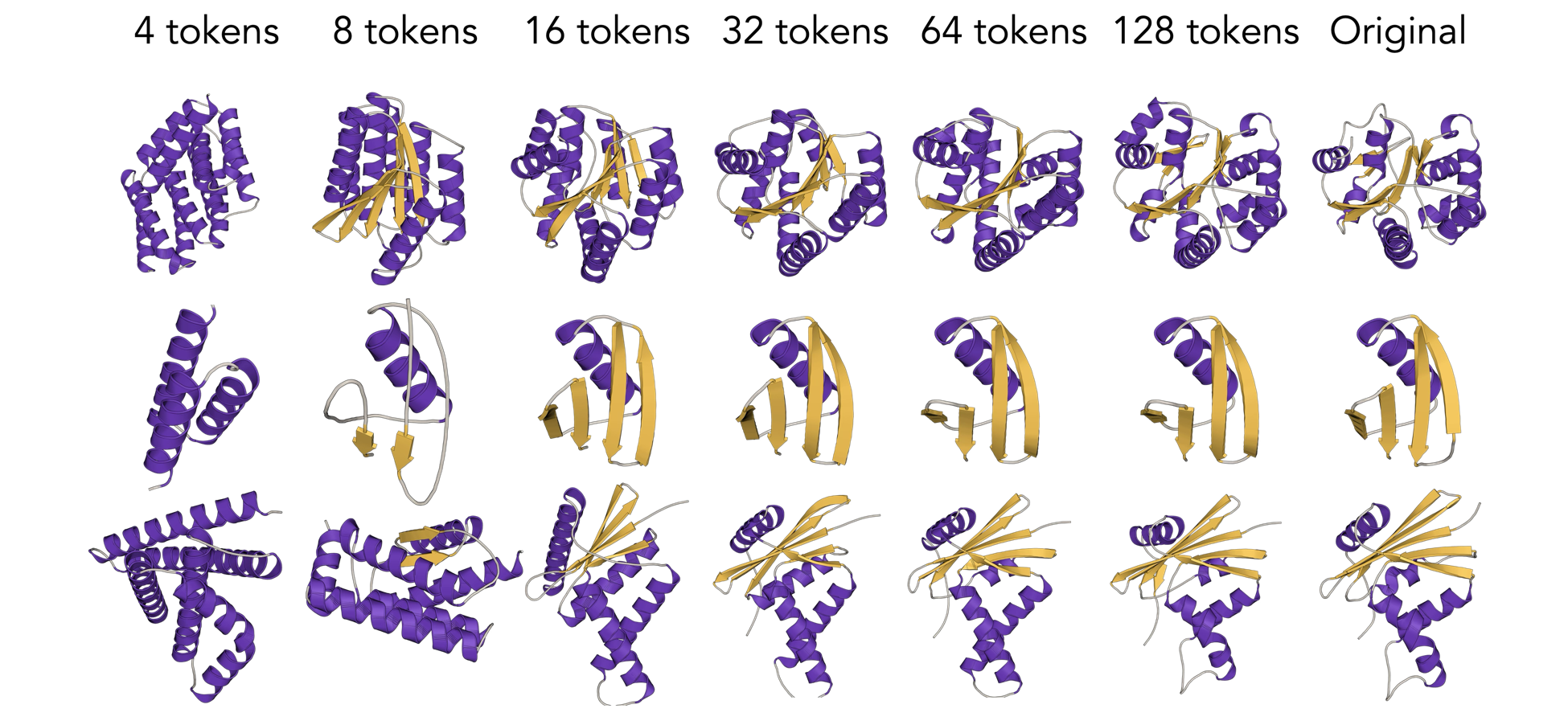
We like this approach for a few reasons. First, we can do a sort of low-pass filtering where we throw away later (high frequency) tokens during decoding. One major failure of autoregressive models is error exposure. If you have some small probability of making a mistake at each token, you have a really high probability of making a mistake at some point during decoding. Once you make one mistake, subsequent mistakes get easier and easier to make as you drift further and further out of distribution. By turning a left-to-right autoregressive sequence into a coarse-to-fine hierarchy, we can throw away tokens likely to contain missamplings. You can't do this with normal tokens because every token corresponds to some region of the protein; if you throw away any part, you'll shorten the protein.
The shift from local+equally weighted tokens to global+adaptive tokens lets us do a bunch of interesting stuff related to inference (e.g., anneal temperature during a rollout). This gives us a lot of improvements in the quality of our generated proteins. See the table below: the metric is designability, and it tracks what fraction of generated proteins are likely to be synthesizable in a lab (so the best you can achieve is 1.0).
| Method | Des. ↑ | scRMSD ↓ | Div. ↓ | Nov. ↓ |
|---|---|---|---|---|
| DPLM2 | 0.486 | 3.31 | 0.300 | 0.615 |
| ESM3 (32 steps) | 0.270 | 17.51 | 0.535 | 0.804 |
| ESM3 (256 steps) | 0.258 | 15.72 | 0.552 | 0.781 |
| DPLM (AR) | 0.320 | 8.99 | 0.297 | 0.656 |
| ESM (AR) | 0.520 | 4.25 | 0.275 | 0.615 |
| Kanzi | 0.562 | 3.78 | 0.517 | 0.779 |
| Kanzi (best-of-N) | 0.617 | 3.65 | 0.511 | 0.786 |
| Ours | 0.871 | 1.352 | 0.395 | 0.774 |
A common usecase for protein models is to use them as a starting point to fine-tune using small amounts of in house data. When you don't have much data, you train a small MLP. But proteins are different lengths, so usually you have a bunch of different vectors (one for each block in the chain). To get a single vector out, most people mean-pool (add all the vectors up and divide by the number of vectors). This is a strange thing to do, but we do so because we don't have a better way. It also doesn't work that well, since you're squashing all that information down. Since our tokens are adaptive, though, we can just take a fixed number of tokens. We test how well we can classify proteins into different folds below -- you can see we do a lot better than existing approaches!
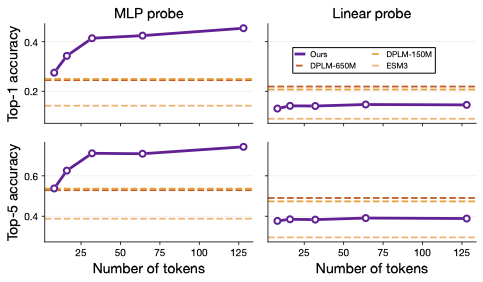
The verdict is still out on whether tokenization is the "correct" way to encode proteins. Unlike in images where the sheer size mandates some form of downpooling, proteins and biological structures don't need much compression. Having said that, the ability to compress proteins according to their genuine information content (as opposed to how long they happen to be) is valuable. We think the techniques we've developed here are going to be useful as we scale to models that are capable of broader and more complex biological tasks.